The Food and Drug administration is responsible for approving prescription and non-prescription drugs for use. This is done through one of the six centers in the FDA called the Center for Drug Evaluation and Research. The other centers are responsible for medical and radiology devices, food and cosmetics, biologic agents, veterinary drugs and tobacco products. Since 1992 over 1,000 new prescription drugs have come to market. So far in 2016 there have been 6 new drugs and in 2015 there were 45. Over the counter medications are handled differently than prescription medicatons. In those cases the FDA only reviews the active ingredients. There are over 30,000 non-prescription drugs on the market.
The table below shows the process, cost and time required for new prescription drug approval. Compounds that potentially have pharmacologic effect are studied in animal models. If they prove promising then an investigational new drug (IND) application is submitted. The FDA has 30 days to review the application and, if needed, put a “clinical hold” on the process to discuss concerns with the applicant. An IND is required to do phase I studies in humans. Phase I studies determine if the compound is safe in humans, side effects and the metabolism and excretion mechanisms. Typically 20-80 healthy volunteers are used.
An Overview of the Drug Development Process
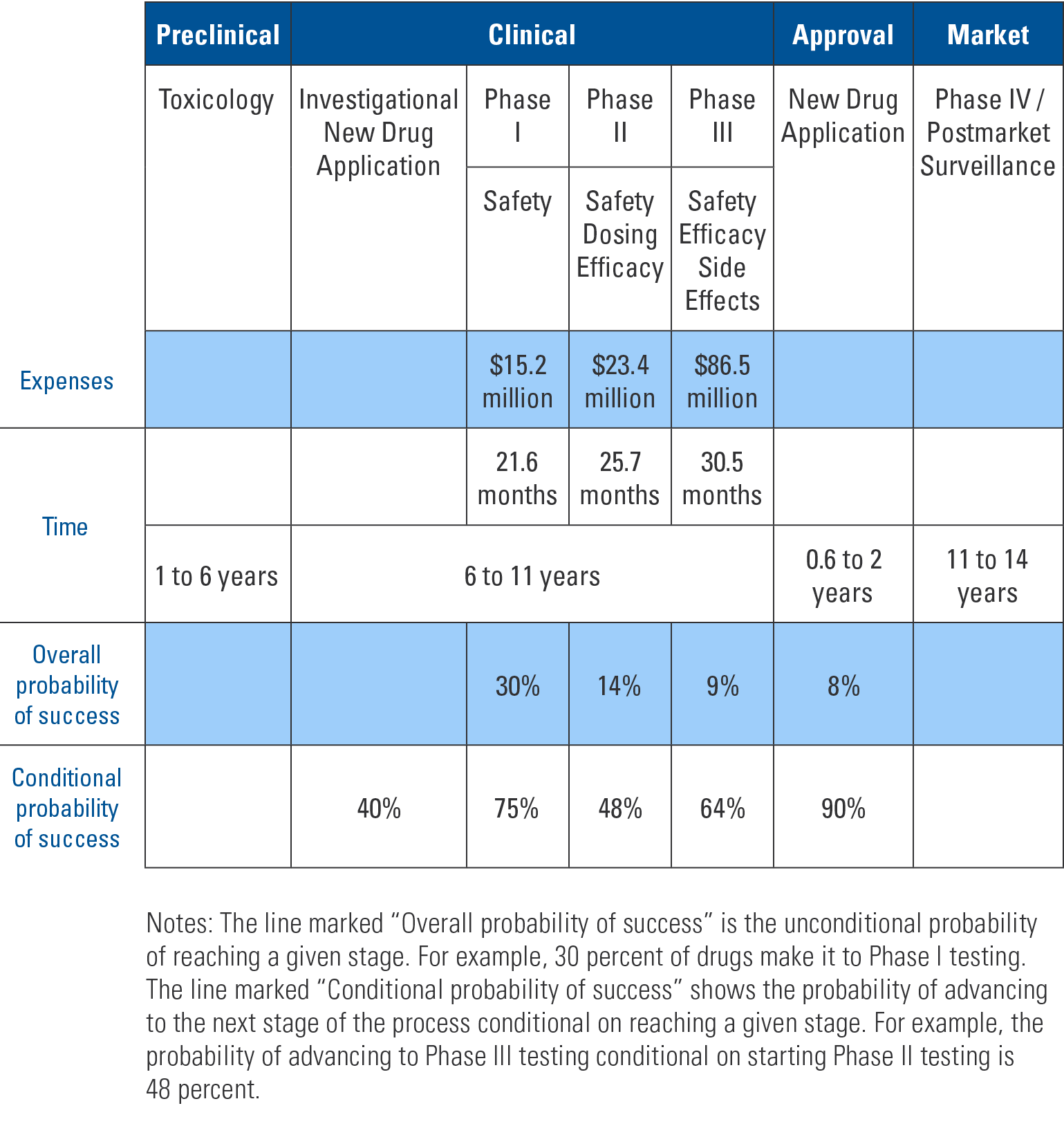
Additionally, before any clinical trial can be done, each study must be approved by an institutional review board (IRB). The IRB is not overseen by the FDA but rather the institution at which the trial is to be performed. The panels are comprised of medical and lay people. They review the studies to ensure safety to participants and that adequate informed consent is obtained.
If phase I studies are positive then phase II studies begin. This phase examines effectiveness of the drug for particular conditions. The drug is compared to either placebo or a similar drug already on the market. These studies involve around 300 participants. Again, if results are positive, then phase III trials are performed. The FDA and drug sponsor agree on large-scale studies to determine safety and effectiveness. Typically trials consist of a few hundred to 3,000 people.
–
“Once a drug is approved it must also undergo phase IV post-marketing surveillance. This is to ensure that when used on a wider population the drug does not cause previously unfound side effects and further verify efficacy.”
After successful phase III studies, a new drug application (NDA) is submitted to the FDA. This application reviews manufacturing specifications, drug stability and bioavailability for each of the dosage forms. Also reviewed are results of toxicological studies and the packaging and labeling for both consumers and prescribers. The goal of the FDA is to decide within 60 days if the NDA can be filed or is incomplete. If filed, the application with all previous studies is sent to committee for final decision. Inspection of the manufacturing facilities is included as part of this NDA process.
Once a drug is approved it must also undergo phase IV post-marketing surveillance. This is to ensure that when used on a wider population the drug does not cause previously unfound side effects and further verify efficacy.
As shown in the table, most compounds do not make it past initial animal studies and even those that do face a steep uphill battle to pass subsequent stages. Even with such rigorous pre-market testing some approved medications are shown to cause harm or not be effective in phase IV surveillance and are withdrawn from the market.